
Leveraging genetic sequencing data of the protein-coding portion of the human genome from more than 60,000 individuals, we found 72 genes to be associated with autism spectrum disorder (ASD) at a stringent threshold of less than 0.1% false discovery rate (FDR). The bulk of the evidence for these implicated genes were contributed by de novo variants, variants arising newly within the offspring that were not present in either parent. We were also able to observe that some genes were preferentially enriched for certain genetic mutation types, such as protein truncating variants or missense variants – observations which could be valuable in unpacking the genetic etiology of ASD. Furthermore, we also integrated an additional 90,000 individuals from the Deciphering Developmental Disorders (DDD) project, a cohort where individuals were ascertained for developmental disorders (DD), and found 373 genes associated with a general neurodevelopmental (NDD) phenotype at a FDR less than 0.1%. Additionally, single-cell data revealed that genes predominantly associated with ASD were enriched in more mature cell types relative to the enrichment of DD-predominant genes, consistent with DD-predominant genes being expressed earlier in development.
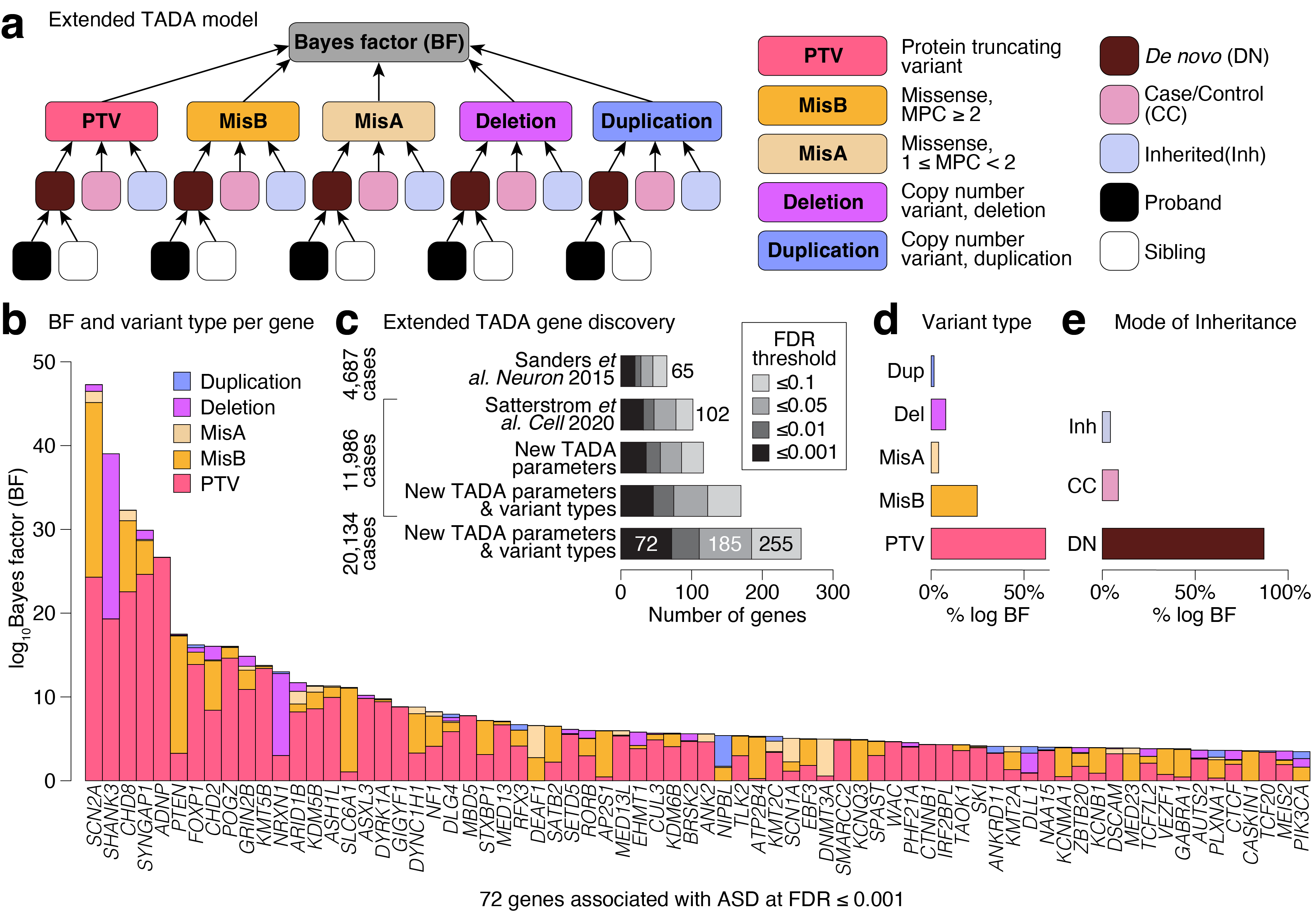
Integrating variant types and inheritance classes significantly boosts association power and reveals mutational biases within candidate genes. a, Our new implementation of the TADA model included de novo, case/control, and rare inherited modules for each variant type: PTV, MisB, MisA, deletion, and duplication. We leveraged information from ASD probands as well as unaffected siblings in evaluating the effect of de novo variants. b, The evidence of ASD association contributed by each variant type for each of the 72 ASD genes with FDR ≤ 0.001. Some genes were predominantly associated with missense variants and duplications (e.g., PTEN, SLC6A1), suggesting mechanisms other than haploinsufficiency. c, Applying TADA to our aggregated ASD dataset yielded 72 genes at FDR ≤ 0.001, compared to 32 and 19 genes at the same threshold in previous studies on a subset of the samples (Satterstrom et al. 2020 and Sanders et al. 2015, respectively). Our expanded TADA model improved the integration of available evidence of association and increased gene discovery at equivalent statistical thresholds on the same datasets. d-e, We quantified the relative contribution of variant class and mode of inheritance to these 72 ASD-associated genes, demonstrating that de novo PTVs and MisB variants represented the strongest contributions to the association signals.
Abbreviations: BF: Bayes factor; PTV: protein truncating variant; MisB: missense variants with MPC score ≥2; MisA: missense variants with MPC score ≥1 and <2; Del: deletion CNV; Dup: duplication CNV; Inh: inherited; CC: case/control; DN: de novo.
Statistical tests: b, Extended TADA model.
Full Article: link
Research Briefing: link