
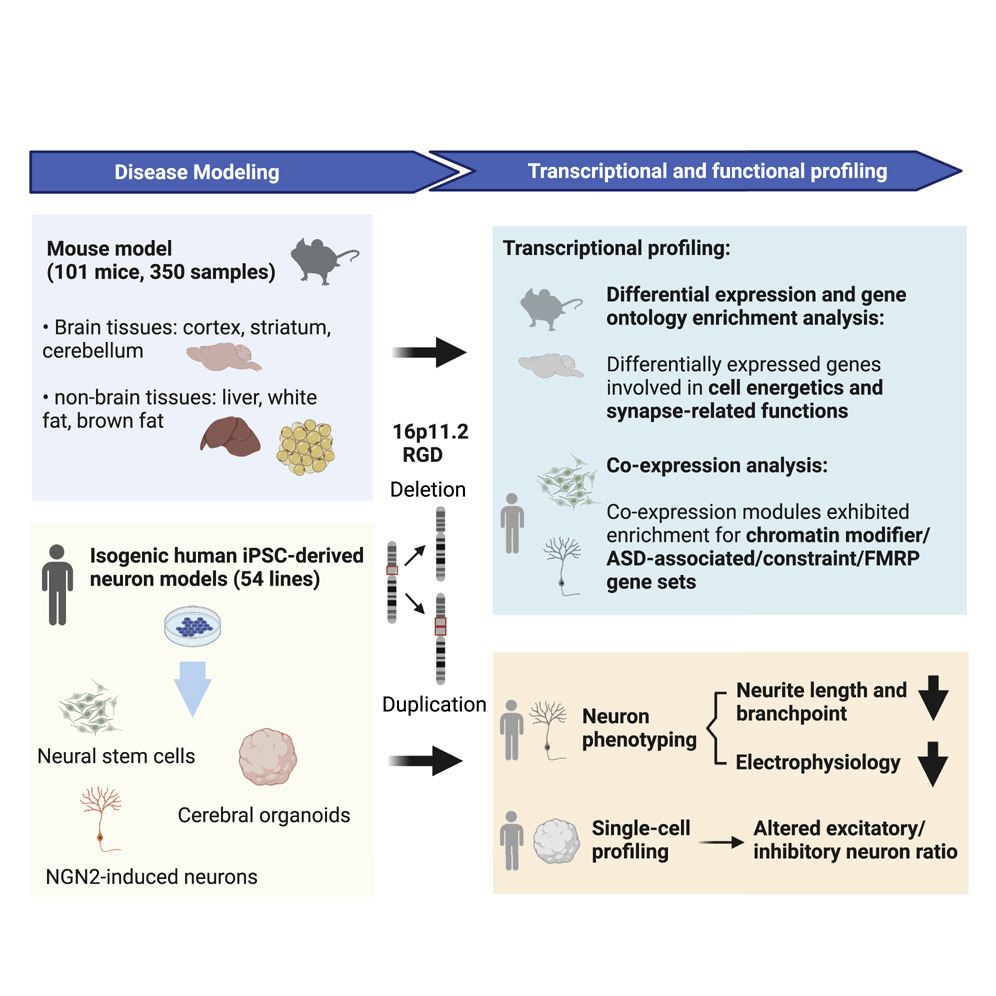
Recurrent and reciprocal copy number variations (CNVs) pose a substantial risk for neurodevelopmental and neuropsychiatric disorders. One example of such CNVs is recurrent deletion and duplication of an ~ 743 kb genomic segment of chromosome 16p11.2, which consists of an ~ 593 kb unique segment encompassing 27 protein-coding genes and an ~ 150 kb flanking segmental duplication encompassing 4 protein-coding gene paralogues. 16p11.2 deletion and duplication have been associated with intellectual disability, autism spectrum disorder, schizophrenia, and an array of anthropometric traits, including obesity. However, how copy number variants at this locus cause these effects is not well understood. To address this challenge, we conducted a multi-omics study, performing transcriptome profiling of 350 samples from three brain tissues (cerebellum, cortex, striatum) and three non-brain tissues (brown fat, liver, white fat) in 16p11.2 mouse models harboring the syntenic 7qF3 CNVs, as well as transcriptional and cellular profiling of 54 isogenic human neural stem cell, induced neuron and cerebral organoid models of CRISPR/Cas9 engineered 16p11.2 CNVs. Our main findings are summarized as below:
1) 16p11.2 deletion and duplication disrupted both tissue-specific and shared biological pathways.
2) 16p11.2 CNVs caused shorter neurites and reduced electrical activity in human cellular models.
3) Single-cell RNA sequencing characterization of human cerebral organoids revealed an altered cellular composition with an excitatory/inhibitory imbalance.
Overall, these findings suggested that the effects of 16p11.2 CNVs on genome-wide gene expression and cellular function are highly dependent on the variant dosage and cellular context.
Full text can be found here.
New! Tai et al. utilize multiple neuronal models to identify tissue- & cell-type specific signatures of 16p11.2 genomic disorders.https://t.co/exol6pYcNH
— AJHG (@AJHGNews) September 23, 2022