
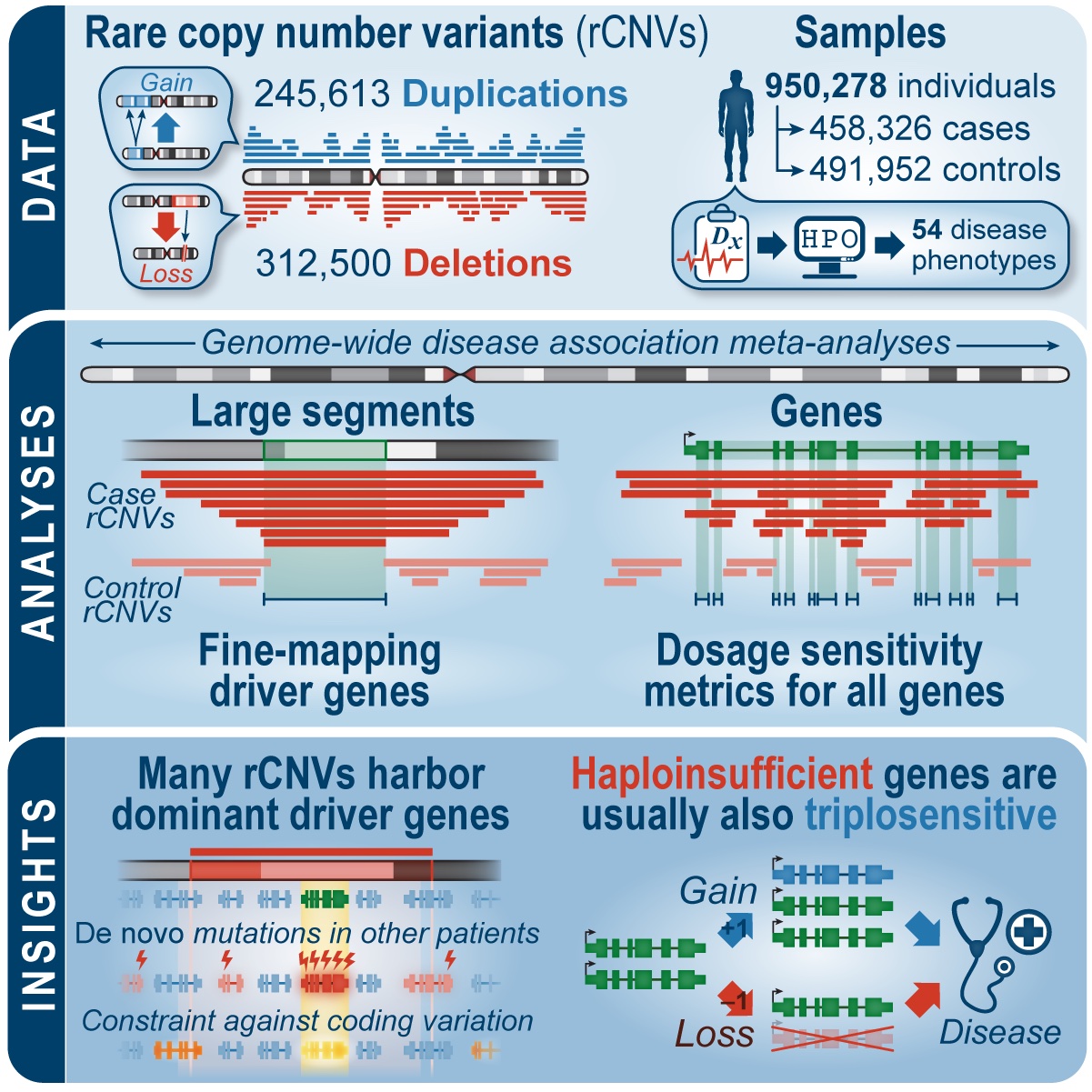
 Large copy number variants (CNVs)—deletions or duplications of more than 100,000 DNA nucleotides—are collectively one of the strongest risk factors for abnormal neurodevelopment. However, interpreting CNVs in clinical genetic practice is challenging due to the lack of standardized resources for predicting the impact of CNVs on individual genes. By harmonizing and jointly analyzing genetic data from nearly one-million individuals, we produced a genome-wide catalog of associations between CNVs and 54 different diseases. We also used a machine learning approach to predict the effects of CNVs on all genes in the human genome, yielding a prediction of 2,987 genes intolerant to loss of one copy (haploinsufficient) and 1,559 genes intolerant to gain of a copy (triplosensitive). These resources will greatly improve interpretation of CNVs in human genetic research and diagnostic screening.
Large copy number variants (CNVs)—deletions or duplications of more than 100,000 DNA nucleotides—are collectively one of the strongest risk factors for abnormal neurodevelopment. However, interpreting CNVs in clinical genetic practice is challenging due to the lack of standardized resources for predicting the impact of CNVs on individual genes. By harmonizing and jointly analyzing genetic data from nearly one-million individuals, we produced a genome-wide catalog of associations between CNVs and 54 different diseases. We also used a machine learning approach to predict the effects of CNVs on all genes in the human genome, yielding a prediction of 2,987 genes intolerant to loss of one copy (haploinsufficient) and 1,559 genes intolerant to gain of a copy (triplosensitive). These resources will greatly improve interpretation of CNVs in human genetic research and diagnostic screening.
Full test can be found here.
Featured article on Spectrum found here.