
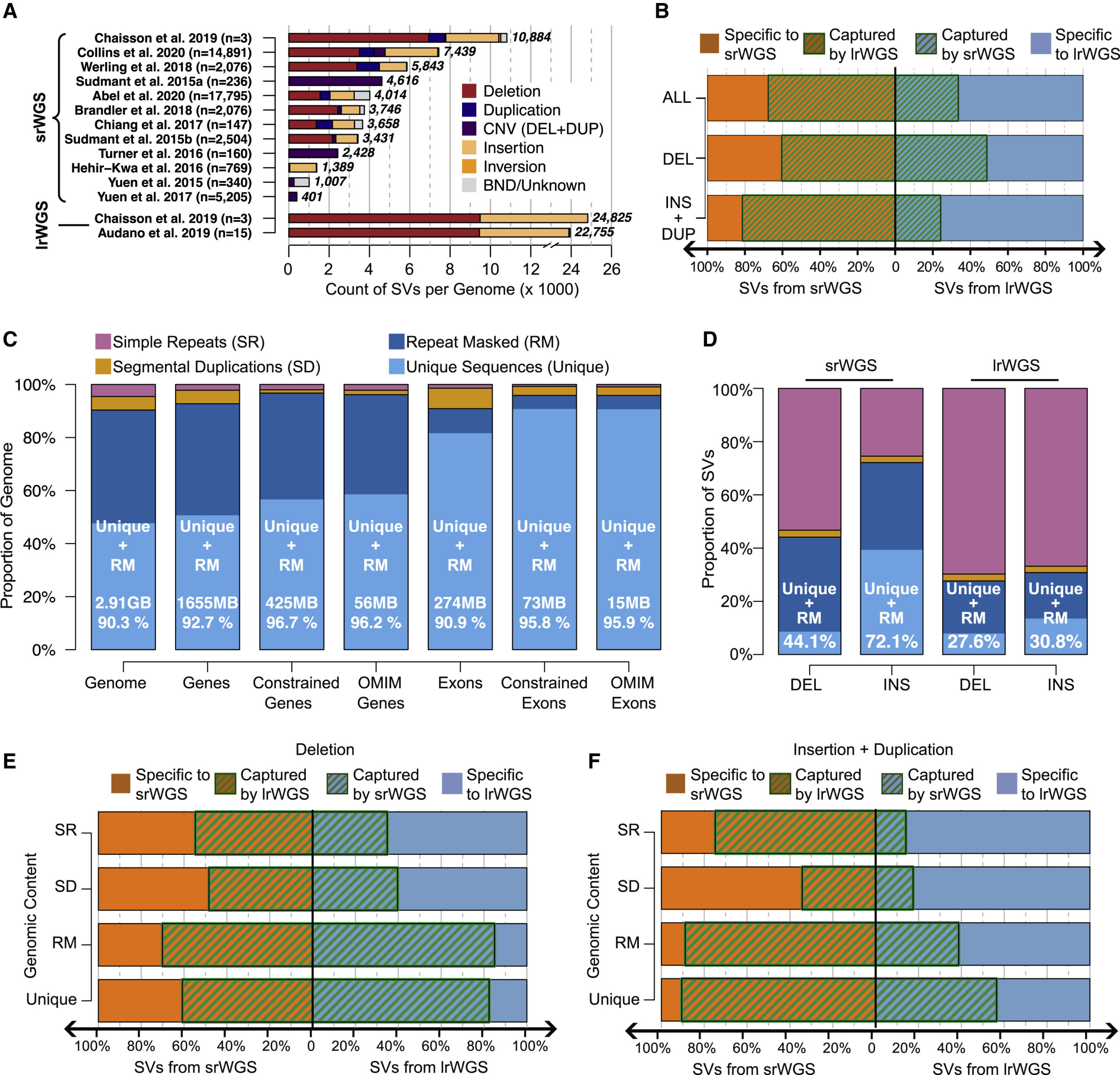
In this study, we conducted in-depth analyses to evaluate the power of different sequencing technologies, including long-read and short-read whole genome sequencing (WGS), in discovering genomic structural variation (SV). Our study revealed that albeit the over two-fold increase in the power of SV discovery by long-read sequences when compared against short reads, vast majority of the difference locate within the highly repetitive genomic regions such as segmental duplicates and simple repeats, which accumulatively consist of <10% of the genome and harbor <5% of the currently annotated coding sequences. While in the rest >90% of genomic regions, where >95% coding sequences are at, performances of long and short reads are comparable. Based upon this study, we estimate current large-scale international initiatives are poised to provide exciting new insights into the 90% of the annotated reference genome that encompasses most known genic sequence. Meanwhile, our analyses also confirmed that assembly-based long-read WGS methods will access regions of the genome that were previously intractable to conventional technologies and long reads. We anticipate that advances in long-read technologies, and associated analytic approaches, will provide significant long-term value in expanding the catalog of functional variation associated with insertions, mobile elements, and the most challenging sequence features in the human genome.
Full text found here.